

Abstract
Background
The clinical phenotype and complications of sickle cell disease (SCD) are heterogeneous and disease outcomes are influenced by both genetic and non-genetic factors, including geography, socioeconomic and educational status and access to health care. These factors serve a role in the quality of life and overall survival in the SCD population. Prior studies have sought to characterize the genotype-phenotype relationship and geographic clustering of SCD with distinct clinical differences found between the African and Asian SCD subtypes. The Central Missouri SCD Cohort (MU-SCD Cohort) has a heterogeneous population of SCD patients who seek medical care from University of Missouri. The clinical characteristics and frequency of SCD-related complications in this group have not yet been defined. This is a retrospective cohort analysis of patients HbSS, HbSC, and HbS/b-thalassemia in central Missouri, ranging in age from the first through the sixth decade of life. We hypothesize that SCD patients from central Missouri, with its mix of African American, Congolese, Nigerian, Kenyan, Southeast Asian and other refugee populations, will have clinical complications distinct from the previously described North American Sickle Cell Disease series and needs further characterization.
Objective
To survey the SCD phenotype by evaluating complications of patients in the MU-Sickle Cell Disease Cohort.
Methods
We retrospectively reviewed clinical and laboratory data for pediatric and adult SCD patients who were treated at the University of Missouri between 2002 to 2018. Prevalence of major vaso-occlusive, hemolytic and renal complications by decade was estimated.
Results
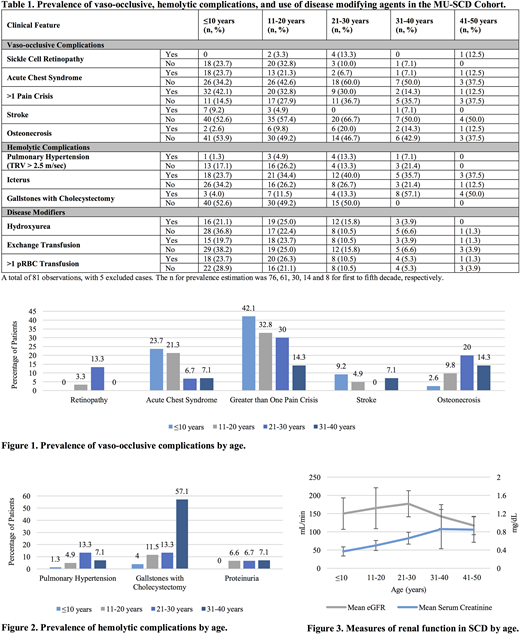
A total of 81 patients were reviewed, with five excluded due to insufficient clinical data. The sample size for prevalence estimation was 76, 61, 30, 14 and 8 for first to fifth decade, respectively. In this cohort, 60.5% were male with average age of 21.2 years. Genotypes represented included 52.6% HbSS, 38.2% HbSC, and 7.9% HbS/b-thalassemia. A total of 94.7% of patients identified as African American. Clinical features of vaso-occlusive complications predominated in the first two decades of life. The frequency of acute chest syndrome for patients ages ≤10 years and 11-20 years of age were 23.7% and 21.3%, respectively (Table 1). Seven children (9.2%) developed overt stroke by age 10 years. Greater than one vaso-occlusive pain crisis occurred in 46% of patients in the first decade of life and thereafter declined with age (Figure 1). In contrast, the MU-SCD Cohort exhibited increasing frequency of hemolytic complications with advancing age. The peak prevalence of pulmonary hypertension occurred between the ages of 21-30 years (13.3%). In congruence, only 4% of patients aged ≤10 years were diagnosed with gallstones requiring cholecystectomy, which increased to 57.1% at age 31-40 years. Reticulocytopenia was noted in the fourth decade with further decline in the fifth decade of life (Figure 2). Renal evaluation revealed an average estimated glomerular filtration rate (eGFR) of 149.9 ± 43 in patients ≤10 years, with progressive decline to 117.3 ± 25 in SCD patients aged 41-50 years (Figure 3). The average serum creatinine demonstrated an increasing trend with advancing age. There was a prevalence of persistent proteinuria in patients older than 11 years. Disease modifying agents were assessed, in which only 21% of patients ≤10 years, 25% of patients aged 11-20 years, and 15.8% of patients aged 21-30 years received therapy with hydroxyurea (Table 1). The use of exchange transfusion or packed red blood cell (pRBC) transfusion for anemia was predominant in SCD patients aged ≤20 years.
Conclusions
This is the first report describing prevalence of SCD-related complications in the MU-SCD Cohort. We identified this population to have an increasing frequency of hemolytic complications and sickle cell nephropathy with advancing age. Onset of persistent proteinuria occurred in the second decade of life, followed by renal insufficiency or end stage renal disease in subsequent decades. As previously demonstrated in the Cooperative Study of Sickle Cell Disease and the Jamaican SCD Cohort study, renal insufficiency was a significant risk factor for early mortality. Further studies are required for identification of biomarkers and institution of early intervention strategies to prevent end-organ damage and decrease mortality in SCD.
No relevant conflicts of interest to declare.

